
By Griffiths, S.C., Schwab, R.A., El Omari, K. et al.
Excerpt from article published in Nature Communication, 12, 7171 (2021). https://doi.org/10.1038/s41467-021-27475-2
Editor’s Highlights
- The Hedgehog (HH) morphogen pathway fulfills crucial functions in growth and morphogenesis, whilst dysregulation leads to developmental disorders and cancer.
- Hedgehog-Interacting Protein (HHIP), the only reported secreted inhibitor of Sonic Hedgehog (SHH) signaling, binds directly to SHH with high nanomolar affinity, sequestering SHH.
- The C-terminal domain of HHIP is composed of a β-propeller and two EGF repeats (HHIP-C).
- The secreted N-terminal domain of SHH (ShhN) is generated from a 45 kDa precursor, undergoing intein-based cleavage to couple an esterified cholesterol molecule to the C-terminus.
- The N-terminal attachment of a palmitoyl moiety produces the fully active lipid-modified signaling ligand (palmitoylated and cholesteroylated ShhN; pShhNc)
- The immunoglobulin superfamily members CDO and BOC bind directly to SHH via the conserved interface involving the pShhNc metal-binding sites.
- Insertion of the SHH palmitoyl and cholesteryl moieties into the ectodomain of PTCH1-B blocks a proposed conduit for cholesterol/sterol transport.
- HHIP-C and PTCH1-A compete for the SHH metal-binding site, which also overlaps with the binding sites for co-receptors CDO, BOC and GAS1.
- HHIP regulates HH signalling. HHIP-C binds to the SHH metal-containing site some 100-fold tighter compared to CDO and PTCH1, and thus could outcompete both CDO and PTCH1 from this site.
-
Aberrant expression of SHH has been linked to the initiation and progression of numerous cancers, and HH inhibitors targeting SMO are in the clinic against basal cell carcinomas.
Abstract
Hedgehog (HH) morphogen signalling, crucial for cell growth and tissue patterning in animals, is initiated by the binding of dually lipidated HH ligands to cell surface receptors. Hedgehog-Interacting Protein (HHIP), the only reported secreted inhibitor of Sonic Hedgehog (SHH) signalling, binds directly to SHH with high nanomolar affinity, sequestering SHH. Here, we report the structure of the HHIP N-terminal domain (HHIP-N) in complex with a glycosaminoglycan (GAG). HHIP-N displays a unique bipartite fold with a GAG-binding domain alongside a Cysteine Rich Domain (CRD). We show that HHIP-N is required to convey full HHIP inhibitory function, likely by interacting with the cholesterol moiety covalently linked to HH ligands, thereby preventing this SHH-attached cholesterol from binding to the HH receptor Patched (PTCH1). We also present the structure of the HHIP C-terminal domain in complex with the GAG heparin. Heparin can bind to both HHIP-N and HHIP-C, thereby inducing clustering at the cell surface and generating a high-avidity platform for SHH sequestration and inhibition. Our data suggest a multimodal mechanism, in which HHIP can bind two specific sites on the SHH morphogen, alongside multiple GAG interactions, to inhibit SHH signalling.
Introduction
The Hedgehog (HH) morphogen pathway fulfils crucial functions in growth and morphogenesis, whilst dysregulation leads to developmental disorders and cancer1,2,3,4. The secreted N-terminal domain of SHH (ShhN) is generated from a 45 kDa precursor, undergoing intein-based cleavage to couple an esterified cholesterol molecule to the C-terminus5. A subsequent step involves the N-terminal attachment of a palmitoyl moiety to produce the fully active lipid-modified signalling ligand (palmitoylated and cholesteroylated ShhN; pShhNc)6. Signalling is activated by binding of pShhNc to the extracellular domains of transmembrane protein PTCH1. Recent structural studies show that PTCH1 and pShhNc form a 2:1 complex, with one molecule of PTCH1 engaging pShhNc at a conserved high-affinity interface involving the conserved SHH zinc- and calcium-binding sites (“protein-protein interface”), and the other at the terminal SHH-palmitoyl and -cholesteryl moieties (“lipid interface”)7,8,9,10,11,12. The PTCH1:pShhNc interaction releases inhibition of the G-protein coupled receptor Smoothened (SMO), which ultimately results in activation of target genes via the GLI transcription factors2. When no HH ligand is present, PTCH1 constitutively inhibits SMO signalling, potentially by preventing access to cholesterol or a similar sterol molecule10,13,14,15,16.
Extracellular distribution of pShhNc is key to the activation of correct signalling responses. This is controlled by a combination of co-receptor signalling17, the glycosaminoglycan (GAG) chains of heparan sulphate proteoglycans (HSPGs)18,19 and the assembly of pShhNc into multimers20,21. The HH pathway is modular, with several other essential cell surface receptors alongside PTCH1. For example, the immunoglobulin superfamily members CDO and BOC bind directly to SHH via the conserved interface involving the pShhNc metal-binding sites22,23,24,25. The metal-binding sites are also crucial for SHH interactions with the vertebrate-specific HH antagonist Hedgehog-Interacting Protein (HHIP)26,27. HHIP is the only secreted inhibitor of HH signalling28,29,30, essential for the development of the lung31, cartilage32 and brain33. HHIP downregulation is associated with HH-dependent tumourigenesis34 and variants at the HHIP locus are linked to Chronic Obstructive Pulmonary Disease (COPD), one of the most common devastating lung diseases in humans35. HHIP is composed of an N-terminal domain (HHIP-N) that shows weak sequence homology to the cysteine-rich domain (CRD) superfamily36, typically involved in small molecule-binding (Fig. 1a). The C-terminal domain of HHIP is composed of a β-propeller and two EGF repeats (HHIP-C). We and others previously determined structures of HHIP-C in complex with human HH ligands ShhN and DhhN, respectively26,27. HHIP utilises a loop inserted into blade 3 of its β-propeller to bind to the HH metal-binding site by directly coordinating the Zn ion. This suggests that HHIP inhibits HH function by sequestering the HH morphogen, and acts as a decoy receptor. Our recent study28 identified a role for HHIP—high affinity interaction with the GAG chains of heparan sulphate proteoglycans (HSPGs), as well as uncovering a cluster of residues in HHIP-N involved in this process. However, the role of the HHIP-N CRD and its potential small molecule binding properties remained elusive.

In this work, we use a combination of structural, biophysical and cellular studies to characterise the HHIP N-terminal region, revealing an unexpected GAG binding domain and a CRD with a small molecule-binding fold. We show that HHIP-N is necessary to convey they full signal inhibition by HHIP in response to pShhNc, and that the purified CRD binds to a mimic of the cholesteroylated HH C-terminus. Importantly, we also identify and structurally characterise two distinct GAG binding sites within HHIP-N and HHIP-C, respectively, and show that HHIP-C assembles into large HHIP-GAG oligomers. Our results reveal that HHIP uses a modular mechanism for SHH inhibition. HHIP targets both the SHH metal-binding and lipid-modification sites recognised by PTCH1, while potentially staying localized on the cell surface via HHIP-GAG interactions.
Results
Structure of the HHIP N-terminal domain reveals a CRD fold
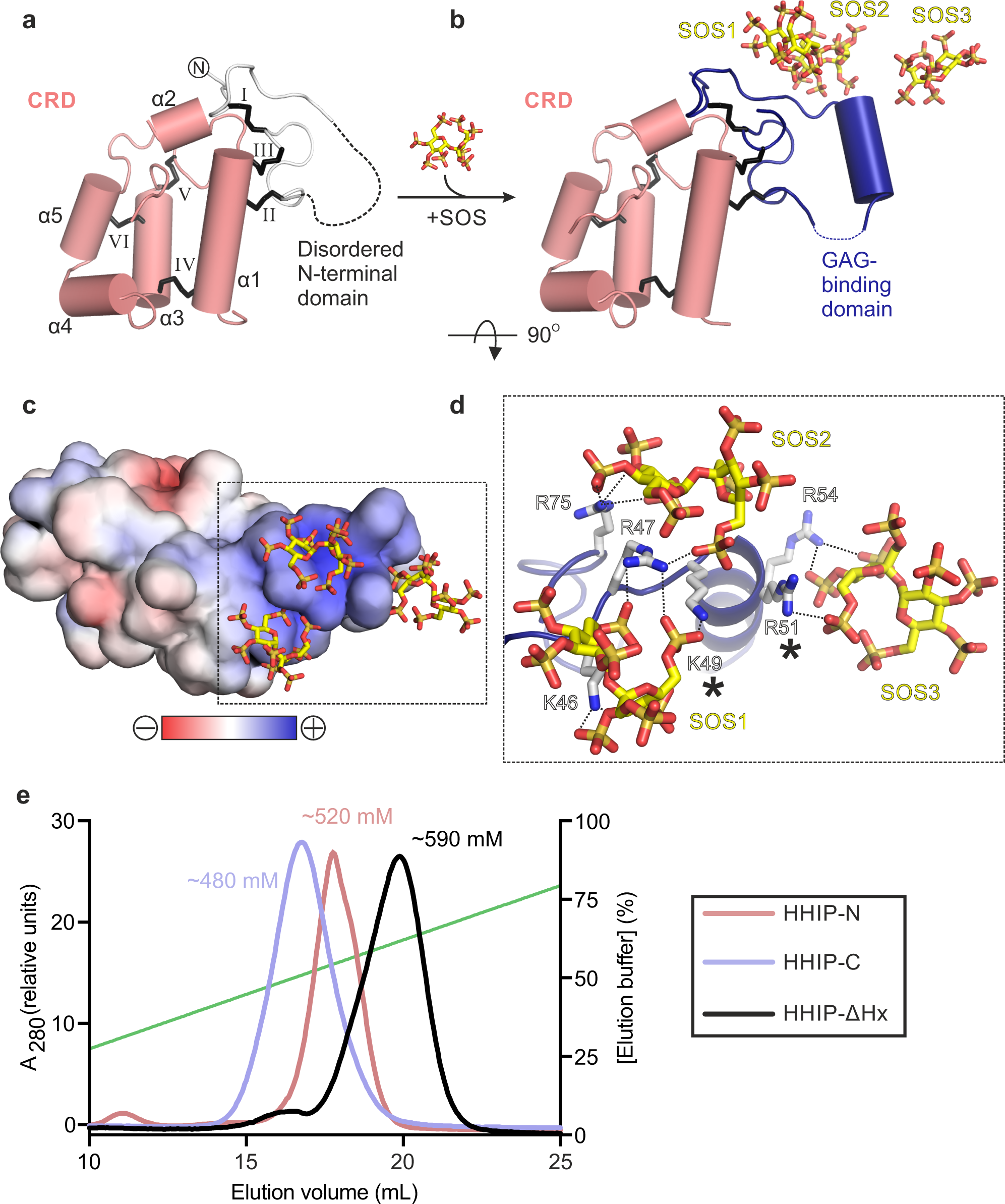
We expressed the N-terminal domain of HHIP (HHIP-N) using mammalian expression in HEK293T cells37 (Fig. 1a). Purified HHIP-N was crystallised in the presence of the GAG mimic sucrose octasulphate (SOS). The structure of the HHIP-N:SOS complex was determined using the single anomalous dispersion (SAD) method from native sulphur atoms. A multi-crystal approach was taken in which 24 data sets were collected from 8 isomorphous crystals, utilising both mini-kappa and inverse beam strategies to maximise the observed anomalous signal (Supplementary Table 1, Supplementary Fig. 1a–d)38,39. This allowed us to determine the structure of HHIP-N in complex with SOS at a resolution of 2.7 Å, with one molecule in the crystallographic asymmetric unit. HHIP-N possesses an elongated globular fold with a unique N-terminal GAG-binding domain (GBD) and a C-terminal CRD (Fig. 1b). HHIP-N is stabilised by a total of 6 disulphide bonds. The GBD of HHIP-N binds SOS, and contains a single helix and flanking loop regions (Fig. 1b, lower panel) that are stabilised by one intra-domain disulphide bond (I: C39-78) and two inter-domain disulphide bonds with helix α1 of the CRD (II: C69-C112 and III: C79-115). The HHIP-N CRD is composed of 5 helices (α1-α5) stabilised by three intra-CRD disulphide bonds (IV: C103-C152, V: C141-179 and VI: C145-168). All disulphide bonds were identified as individual sites in the anomalous difference map, and treatment of these as ‘super-sulphurs’ was vital for substructure determination and structure solution (Fig. 1c)40.
CRDs can bind to various small molecules, including folate (folate receptors; FRα/β)41 and riboflavin (riboflavin-binding protein; RFBP)42, as well as lipids and sterols, such as cholesterol (SMO, NPC1)43,44 and palmitoleic acid (Frizzled; Fz)45. Previously, we performed an evolutionary structural analysis to classify known CRDs into two distinct sub-families, containing ‘pockets’ or ‘grooves’ as ligand-binding elements46. In this context, a ‘pocket’ refers to a CRD in which the ligand-binding site consists of loops that fold over a deep cavity inaccessible to solvent, whereas a ‘groove’ comprises a shallow and elongated solvent-accessible binding cleft, which does not undergo major conformational changes upon ligand binding. We have now included our HHIP-N structure in this analysis and classified it as a member of the pocket-type sub-family, with related structural homologues in the CRDs of NPC1, RFBP and FRα/β (Fig. 1d, Supplementary Table 2). Additionally, this analysis identifies sperm-egg fusion protein JUNO as being a pocket-type CRD which has diverged from typical folate receptors47. The HHIP-N CRD comprises a structural ‘scaffold’ composed of helices a1, a3 and a5 that are stabilised by disulphide bridges, a feature common to all CRD family members (Fig. 1b, Supplementary Fig. 2). In our HHIP-N structure, we observed two stretches of disordered residues, which link the N-terminal domain and CRD helix α1 (DL1), and the loop regions between helices α2 and α3 (DL2), respectively (Fig. 2a). In FRβ, the equivalent regions form the majority of a folate-binding pocket (Fig. 2b), suggesting that DL1 and DL2 very likely occupy a similar region. The modes of ligand binding by pocket-type CRDs are illustrated in Supplementary Fig. 3b, c and Supplementary Fig. 3f, while ligand binding by groove-type CRDs is detailed in Supplementary Fig. 3d-e. Helices analogous to HHIP-N α1-5 form an equivalent ‘scaffold’ in the pocket-type CRDs NPC1, RFBP and JUNO, within which ligand-binding loops are positioned for small molecule binding. A ligand-binding loop comparable to HHIP-N DL1 is also present in the groove-type CRDs of Fz8 and SMO (Supplementary Fig. 3d, e). This structural and evolutionary analysis suggests that loops DL1 and DL2 could potentially form a binding pocket for a physiologically-relevant small molecule ligand within the HHIP-N CRD region.

Next, we tested the function of HHIP-N in a SHH signalling assay, based on measuring the mRNA levels of the HH target gene Gli1 in NIH/3T3 cells. We co-cultured PANC-1 human pancreatic ductal adenocarcinoma cells expressing pShhNc48 to activate the HH pathway. The purified full-length HHIP extracellular domain only lacking the C-terminal helix (residues 39-670, HHIP-∆Hx, Fig. 1a) was able to inhibit HH signalling more efficiently than HHIP-C (Fig. 2c), which lacks HHIP-N but contains the high-affinity SHH binding site26,27. We next asked whether the interaction of HHIP-N with the SHH lipid modifications could account for this increase of inhibition, given the structural similarities with small molecule-binding CRDs. For this, we activated the HH pathway with an N-terminal palmitoylated-SHH peptide (Palm-ShhN15)49, which does not interact with HHIP-C. In this context, HHIP-∆Hx was unable to inhibit signalling (Fig. 2d), excluding a role of the SHH N-terminal palmitoyl moiety in HHIP-N binding. We recently showed that the SHH C-terminal cholesterol moiety is important for SHH-PTCH1 interactions and activation of HH signalling12. Using a similar isothermal titration calorimetry (ITC) assay, we tested whether a PEGylated cholesterol molecule (mimicking the C-terminus of pShhNc) binds to HHIP. PEG-cholesterol bound specifically to HHIP-N (KD = 23 µM) (Fig. 2e), whereas free PEG did not bind to HHIP-N (Fig. 2f). Due to the high heat of dilution when PEG-cholesterol is titrated into buffer, a series of controls were conducted (Supplementary Fig. 4). Thermodynamic signature plots for HHIP-N compare favourably with those obtained for the recently published interaction of PEG-cholesterol with the canonical Hh receptor PTCH112, revealing a high enthalpic contribution to ∆G suggesting a hydrophobic interaction50. This supports that the interaction between PEG-cholesterol and HHIPN is specific. Taken together, the enhancement of HH signalling inhibition by HHIP in the presence of HHIP-N, combined with structural homology to small molecule-binding pocket-type CRDs and observed binding to PEG-cholesterol suggests an interaction between HHIP-N and HH-linked cholesterol. Hence, we propose a model whereby HHIP binds to SHH in a multimodal manner, utilising the SHH metal-binding sites for HHIP-C binding and potentially targeting the cholesterol attachment site for HHIP-N binding. Such an arrangement could be accommodated in a 1:1 SHH-HHIP complex, as HHIP-N and HHIP-C are separated by a 25-residue long, likely flexible linker that could position the SHH-linked cholesterol to interact with HHIP-N and the SHH-metal binding site to interact with HHIP-C within the same molecule (Supplementary Fig. 5).
The HHIP N-terminal domain contains a GAG-binding domain
To shed light onto the previously reported GAG-binding properties of HHIP-N28, we determined structures of apo and SOS-bound HHIP-N to resolution of 2.6 Å and 2.7 Å, respectively (Supplementary Table 1). In the apo HHIP-N structure, the N-terminal GAG-binding domain is disordered, while the CRD region is inherently structured (Fig. 3a). This suggests that interaction of HHIP-N with GAG molecules triggers a transition to a folded state, forming an α-helix between residues 50 and 58. Each HHIP-N molecule interacts with 3 SOS molecules (Fig. 3b, Supplementary Fig. 6). SOS is bound to a positively charged surface localised to the HHIP-N N-terminal domain (Fig. 3c), forming electrostatic interactions with a cluster of 6 basic residues and burying a total surface area of 594 Å2 (Fig. 3d). Only Arg-47 forms hydrogen bonds with more than one SOS molecule; Arg-51 and Arg-54 are the only residues within the N-terminal α-helical residues 50–58 to contact a SOS molecule. Mutagenesis of SOS-interacting basic HHIP residues (marked with asterisks in Fig. 3d) were previously observed to weaken the interaction between HHIP and heparin, validating our observed HHIP-N:SOS interface28. The N-terminal domain of HHIP-N is different compared to other structurally characterised CRDs, having evolved a GAG-binding function in a discrete domain alongside a conserved small molecule-binding fold. Additionally, we report structural insights into the major secondary structural rearrangements of HHIP-N, a GAG-binding protein, upon sugar coordination.
Structure of the HHIP C-terminal domain in complex with heparin
Our previous analysis of HHIP:GAG interactions identified multiple GAG-binding motifs in HHIP, since a construct that contains a deletion of HHIP-N was still able to bind to heparin27. To decipher the GAG-binding properties of HHIP, we conducted binding assays using three domain deletion constructs (Fig. 1a). First, we tested GAG binding using heparin affinity chromatography (Fig. 3e). Both HHIP-C and HHIP-N show binding to a heparin column, albeit with weaker affinity when compared to HHIP-∆Hx. Furthermore, HHIP-N, HHIP-C and HHIP-∆Hx are all cell surface-associated at physiological pH and ionic strength (Supplementary Fig. 7). Taken together, our analysis suggests that both HHIP-N and HHIP-C contribute to the observed affinity for GAG chains.
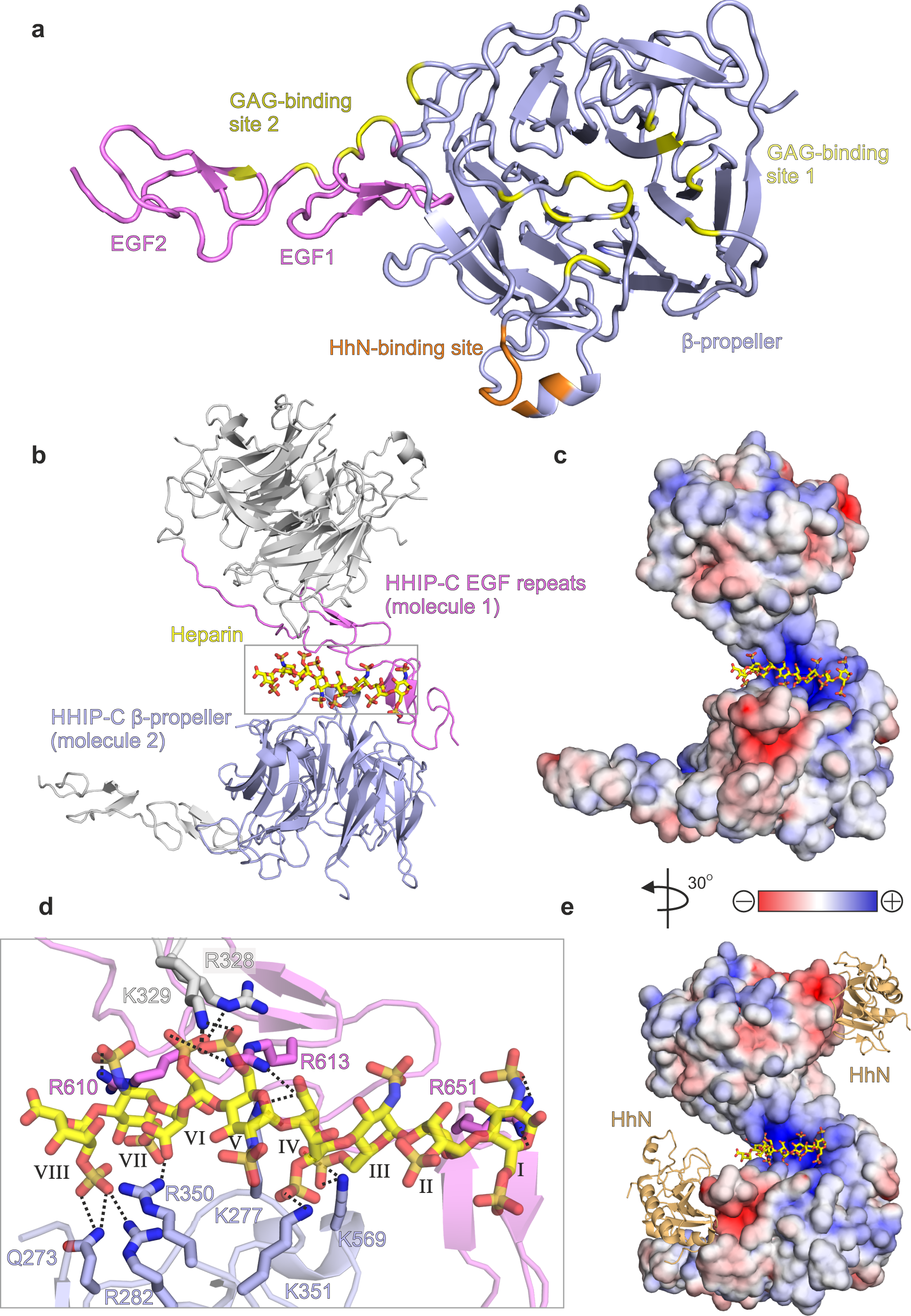
To structurally characterise HHIP-C:GAG interactions, we determined the 2.7 Å resolution crystal structure of HHIP-C in complex with a 30-mer heparin molecule (Supplementary Table 1, Supplementary Fig. 8a). HHIP-C comprises an N-terminal 6-bladed β-propeller and 2 C-terminal EGF repeats, previously identified in the complex of HHIP-C with HhN ligands26,27. The heparin chain contacts two separate clusters of HHIP surface residues (GAG ‘site1’ and GAG ‘site 2’) at either end of the molecule (Fig. 4a, Supplementary Fig. 8). In this arrangement, a HHIP-C anti-parallel dimer coordinates one single heparin chain (Fig. 4b) by using a central positively-charged region formed by residues from both HHIP protomers (Fig. 4c). The HHIP-C chains in the dimer are essentially identical (r.m.s.d. of 0.21 Å for 322 equivalent Cα atoms), and show little structural difference to previously published HHIP-C structures (e.g. r.m.s.d. of 0.31 Å for 331 equivalent Cα atoms, PDB ID. 2WFT26). The heparin backbone displays a right-handed helical structure with roughly 4 sugars per turn, in agreement with previous structural studies51,52. A total of 8 monosaccharides are resolved in the structure, running from a reducing (O1; sugar I) to a non-reducing end (O4; sugar VIII) and forming hydrogen bonds with several polar side chains (Fig. 4d). We also determined the crystal structure of a HHIP-C:SOS complex, which exhibits the equivalent anti-parallel, dimeric HHIP arrangement bound to two SOS molecules coordinated at the same site compared to heparin (Supplementary Table 1, Supplementary Figs. 8b, 9 and 10). In summary, our HHIP-C crystal structures in complex with GAG molecules identify two discrete GAG-binding sites on the surface of HHIP-C, which are distinct and non-overlapping with the HhN-binding interface (Fig. 4e), thus suggesting that both GAG- and HhN-binding can occur simultaneously.
GAG interactions control oligomerisation of HHIP
Our previous study on HHIP:GAG interactions identified low micromolar affinities for the interactions between HHIP-C and the GAGs heparin, heparan sulphate (HS) and chondroitin sulphate (CS)28. To experimentally validate our HHIP-C:GAG complex structures, we generated a HHIP mutant in which 6 positively-charged residues involved in GAG recognition were mutated to glutamate (K277E/R328E/R350E/K569E/R610E/R613E, HHIP-C Glu mutant; Fig. 4d) and analysed binding to GAGs. Using surface plasmon resonance (SPR) with GAGs immobilised on the chip, we observed that the HHIP-C Glu mutant almost completely abolishes binding to heparin, HS and CS, when compared to wild-type (HHIP-C WT) (Fig. 5a). Furthermore, we analysed our observed HHIP-C:GAG interfaces by a combination of mutagenesis and heparin affinity chromatography. In addition to the HHIP-C Glu mutant, we generated glycosylation mutants (Supplementary Fig. 10a). N-linked glycans were inserted into the GAG-interacting surfaces of either the HHIP β-propeller (‘ΔGAG site 1’) or EGF repeats (‘ΔGAG site 2’), as well as combination of both (‘ΔGAG sites 1 + 2’). As expected, all mutants showed reduced heparin binding affinity relative to HHIP-C WT (Supplementary Fig. 11b). Next, we tested the effect of HHIP-C WT or HHIP-C Glu mutant in SHH signalling assays using NIH/3T3 cells (Fig. 5b). We observed that the HHIP-C Glu mutant showed significantly less efficient SHH inhibition. To further probe whether this effect results from loss of GAG-binding, we knocked out EXTL3, a key enzyme in the biosynthesis of cell-surface heparan sulphate (HS) chains (Supplementary Fig. 12a–d)53. We confirmed that single Extl3−/− NIH/3T3 clones lack surface HS chains by showing markedly reduced binding to a scFv antibody (HS20) that is known to recognize multiple HS chains54,55 (Supplementary Fig. 12e). These Extl3−/− clones were responsive to SHH, showing that the machinery required to receive and respond to ligands was unaffected by the loss of HS chains (Supplementary Fig. 12e). Compared to their individual level of SHH activation, wildtype HHIP was a less effective inhibitor in both knock-out cell lines than in their parental cell line. Even more importantly, both HHIP wildtype and Glu mutant inhibited the HH response in the HS-deficient cells to similar degrees, in contrast to the parental cell line where Glu mutant HHIP was a less effective inhibitor (Supplementary Fig. 12e). This demonstrates that the interaction between HHIP-C and GAGs are critical for maximal HH pathway inhibition.
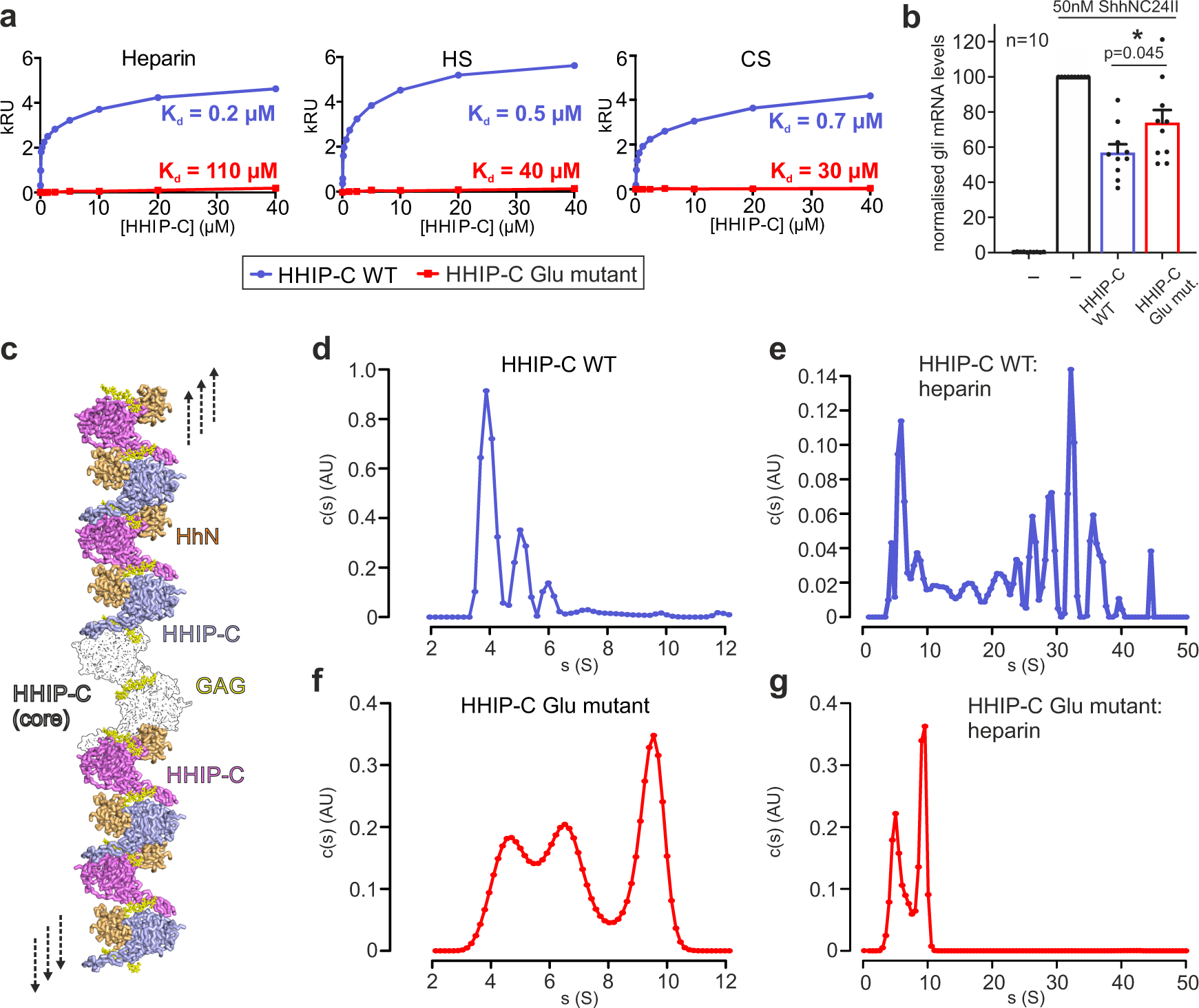
Our HHIP-C:heparin complex structure also suggests that GAGs can mediate the formation of long oligomeric chains of HHIP (Fig. 5c). We observe two HHIP oligomerisation interfaces – an anti-parallel ‘head-to-tail’ heparin-bound HHIP-C dimer (Supplementary Fig. 13a) with a total buried surface area of 603 Å2 (Supplementary Fig. 13a), and a ‘head-to-head’ dimer with a buried surface area of 981 Å2 (Supplementary Fig. 10b) in the crystal. The ‘head-to-tail’ dimer interface is smaller than physiological dimer interfaces and is likely stabilised by the heparin molecule (PISA ΔiG P-value = 0.41, shape complementarity score = 0.75)45,46, while the larger ‘head-to-head’ interface is more likely to be physiological (PISA: ΔiG P-value = 0.43, shape complementarity score: 0.52). Previous studies using multi-angle light scattering (MALS) showed that the apo HHIP-C is monomeric at a concentration of approximately 10 μM26. To test whether oligomers observed in the crystal structure exist in solution at higher concentrations (potentially mimicking the local concentrations at the cell membrane), we performed sedimentation velocity analytical ultracentrifugation experiments (AUC). This revealed a predominantly monomeric apo HHIP-C population (4 S, 53 kDa), with minor populations of dimers (5 S, ~100 kDa) and trimers (6 S, ~150 kDa) also observed (Fig. 5d). As expected, addition of the heparin 30-mer triggers a drastic shift to increased sedimentation rates, and causing the sedimentation of an array of higher-order oligomeric species from 50 kDa up to 6 MDa (4 S–50 S; Fig. 5e). Higher-order oligomer formation in response to the addition of heparin is completely ablated in the HHIP-C Glu mutant (Fig. 5f–g), consistent with GAG-binding induced oligomerisation of HHIP. Taken together, our biophysical and cellular data suggest that HHIP:GAG complexation leads to HHIP clustering, and these assemblies might fine-tune inhibition of the HH signal.
HHIP-N can also bind GAGs with high affinity and despite crystallising as a monomer, forms weak dimers in solution when analysed using MALS (Supplementary Fig. 14a). Interestingly, apo HHIP-N is predominantly monomeric in solution up to 200 µM when studied by AUC (Supplementary Fig. 14b), and can also form oligomers in the presence of heparin (up to a size consistent with tetramers, as shown from c(s, f/f0) plots to analyse molecular weights in solution; Supplementary Fig. 14c). This is consistent with our crystal packing analysis (Supplementary Fig. 4), and suggests that GAGs stabilise a contact between HHIP-N GAG-binding domains in solution to assist oligomerisation (Supplementary Fig. 14d). In conclusion, HHIP is regulated through both protein:protein- and protein:GAG-mediated oligomerisation at several different sites, that can be linked to dynamic modulation of HH signalling.
Discussion
The function of the N-terminal domain of HHIP has remained a long-standing mystery. Previous work in our laboratory and by others showed that HHIP-C binds to the metal-binding site of SHH with high (low nanomolar) affinity, and inhibits HH signalling by functioning as a decoy receptor26. Our structural, biophysical and cellular work reveals that HHIP-N contains a CRD, which acts as an additional module within HHIP to inhibit HH signalling. Moreover, HHIP-N has evolved a structured region engaged in cell surface heparan sulphate proteoglycan binding. Recent pioneering work on the PTCH1:pShhNc interaction has greatly enhanced our understanding of HH signal reception and transduction. PTCH1 and SHH interact in a 2:1 stoichiometry, with one PTCH1 molecule (PTCH1-A) binding to the high-affinity metal-binding site of SHH and the other PTCH1 molecule (PTCH1-B) grasping the two lipid modifications to form the full signalling complex (Fig. 6a). None of these interactions alone is sufficient to fully inactivate PTCH1 function in cellular assays12. This mechanism shows parallels to WNT morphogen signalling, in which WNT interacts with its receptor Frizzled (Fz) with both its covalently-linked palmitoleate (via the Fz-CRD, evolutionarily related to the HHIP-N CRD), and also via a protein-protein interface45. WNT inhibition is, in part, achieved by interactions with secreted Fz-related proteins (sFRPs) (via both palmitoleate- and protein-protein contacts), which act as secreted decoy receptors56. Our structural and functional analysis suggests that HHIP has potentially evolved a similar role, targeting the SHH cholesterol moiety (via HHIP-N) and the metal-binding site (via HHIP-C). Thus, a two-pronged engagement with both protein- and lipid-based interfaces seems to be a common theme in recognition of morphogens.
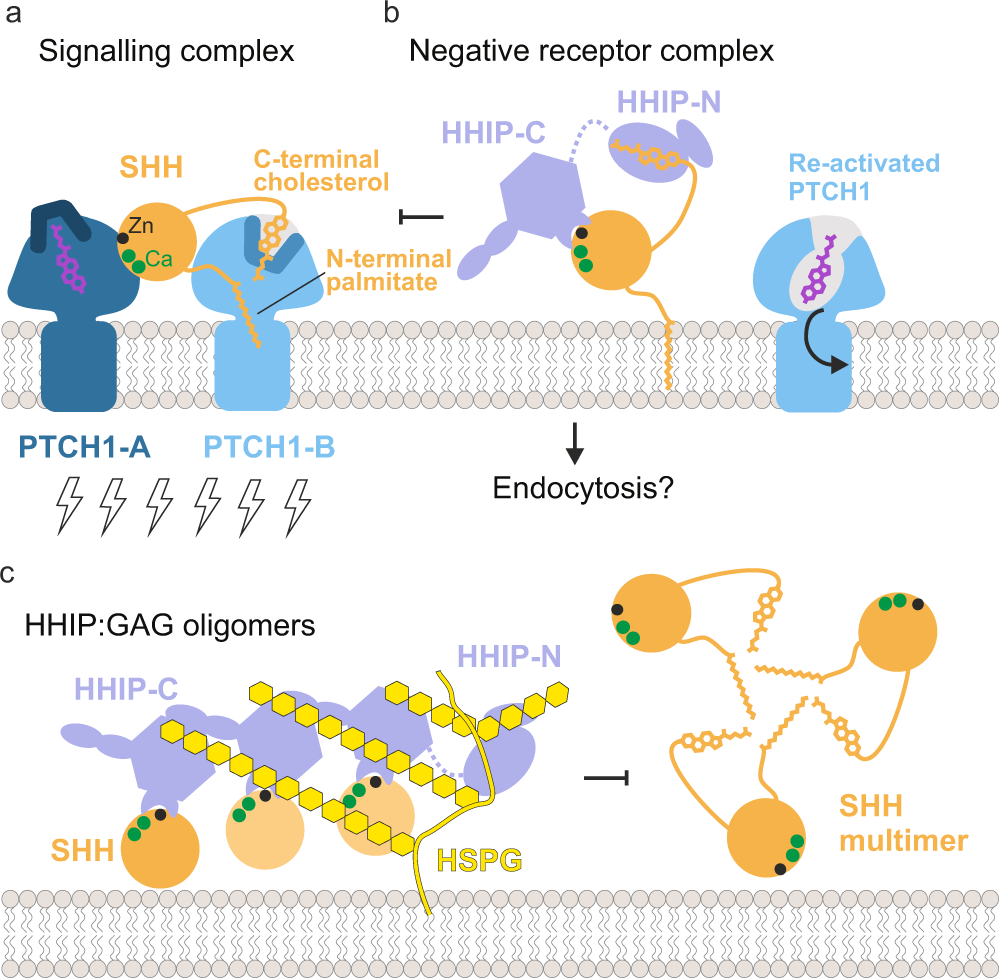
Insertion of the SHH palmitoyl and cholesteryl moieties into the ectodomain of PTCH1-B blocks a proposed conduit for cholesterol/sterol transport57,58. Shielding of the pShhNc cholesterol modification by HHIP-N would be an effective mechanism to release SHH-mediated inhibition and transport function of PTCH1. This is complemented by HHIP-C and PTCH1-A competing for the SHH metal-binding site, which also overlaps with the binding sites for co-receptors CDO, BOC and GAS117,22. These two modes of signal antagonism constitute a fail-safe mechanism by which HHIP regulates HH signalling. HHIP-C binds to the SHH metal-containing site some 100-fold tighter compared to CDO22,26 and PTCH112, and thus could outcompete both CDO and PTCH1 from this site. This could result in the formation of a ‘negative receptor complex,’ facilitating for example SHH endocytosis (Fig. 6b).
Various studies have identified HHIP as the only known secreted diffusible inhibitor of HH signalling28,29,59. We previously showed that HHIP secretion is dependent on interaction with GAGs, and that HHIP can bind to various types of GAGs28. Here, we have delineated the molecular basis for this interaction. Both HHIP-N and HHIP-C contain discrete GAG-binding sites, which combine to modulate cell surface affinity. Together, they constitute additional minor regulatory sites of HH signalling. Cell surface-attached GAGs such as HS and CS organise secreted proteins into gradients, varying local concentrations to enable graded signal activation60. Here, we show that GAGs are able to cluster HHIP-C into large assemblies, linked to the potency of HHIP-mediated HH inhibition (which is further potentiated by HHIP-N). In addition, the HHIP C-terminal helix (residues 671–700) that contributes to cell surface binding via formation of a predicted leucine zipper-type assembly28 may enhance clustering even further, combined with the dimerisation abilities observed for both HHIP-N and HHIP-C. HHIP clustering by GAGs at the cell surface generates a high-avidity platform for SHH binding. This platform might limit diffusion and loss of HHIP into extracellular space and position HHIP to inhibit SHH signalling at the cell surface (Fig. 6c). This process can be further regulated by SHH-mediated internalisation of HHIP29, which potentially can happen at both SHH producing or responding cells. From our structural analysis, we also note that there is a long flexible linker between HHIP-N and HHIP-C, which we previously described as being proteolytically sensitive26. Cleavage of this linker may regulate the overall cell surface affinity of HHIP in vivo. Taken together, our results indicate that HHIP regulation of HH signalling is dynamic, and inhibitory processes are modularised through distinct functionalities within the HHIP N- and C-terminal domains.
Aberrant expression of SHH has been linked to the initiation and progression of numerous cancers, and HH inhibitors targeting SMO are in the clinic against basal cell carcinomas3,4. The SHH-specific mouse antibody 5E1 binds tightly to the SHH metal-binding site, overlapping with HHIP61, and has been shown to inhibit SHH activity in vivo and to reduce tumour growth in a pancreatic cancer mouse model62. HHIP could be used in a similar way, working as an engineered biologic to inhibit HH signalling. Here, we provide the framework for the development of HHIP-based HH inhibitors that can specifically preserve or block the different SHH-receptor interaction sites, and this interaction mode can be further fine-tuned by the GAG-binding properties and resultant HHIP clustering. Various biomedically-important secreted signalling proteins are reported to undergo GAG-dependent clustering63, and drugging such mechanisms could present an unexploited avenue for therapeutic discovery.